Quick start guide
See vignette("authentication") for instructions on
creating your personal API token for accessing data. To begin using
pluto, load the package and log in with your personal API
token:
library(pluto)
pluto_login("<YOUR API TOKEN>")Accessing Seurat objects from Pluto
Single cell RNA-seq in Pluto creates two Seurat objects:
a raw Seurat object, which is created at
the beginning of the single cell RNA-seq preprocessing workflow and has
not been filtered, normalized, integrated, clustered, etc.; and a
final Seurat object, which is the final,
processed Seurat object that results from completing and
accepting a preprocess workflow. Both Seurat objects are
accessible for external scRNA-seq analysis via the pluto
package.
Download Seurat object
To download a Seurat object, provide the experiment ID,
Seurat type ("raw" or "final") to
the pluto_download_seurat_object() function (you can also
provide an optional desired destination file path by setting
dest_filename):
experiment_id <- "PLX037839"
seurat_type <- "raw"
pluto_download_seurat_object(experiment_id, seurat_type)Read Seurat object
To read a Seurat object directly into your R script,
provide the experiment ID and Seurat type ("raw" or
"final") to the pluto_read_seurat_object()
function:
experiment_id <- "PLX037839"
seurat_type <- "raw"
so <- pluto_read_seurat_object(experiment_id, seurat_type)It is important to note that the
pluto_read_seurat_object() function returns different
objects depending on the Seurat type specified. If the
Seurat type is specified as "raw", a Seurat
object will be returned. If the Seurat type is specified as
"final", a list is returned containing the final
Seurat object, obj, with custom cluster
annotations added to the Seurat meta.data and
custom color palettes corresponding to the cluster annotation sets
in-app, colors.
experiment_id <- "PLX037839"
seurat_type <- "final"
final_obj <- pluto_read_seurat_object(experiment_id, seurat_type)
# To access the Seurat object
so <- final_obj$obj
# To access the custom color palettes
custom_palettes <- final_obj$colorsThe Seurat object
The Seurat object, developed by the Satija lab, is
a representation of single-cell expression data for R. The object serves
as a container that contains both data (like the count matrix) and
analysis (like PCA, or clustering results) for a single cell dataset. At
the top level, the Seurat object serves as a collection of
Assay and DimReduc objects, representing
expression data and dimensionality reductions of the expression data,
respectively. Seurat objects also store additional
metadata, both at the cell and feature level. Click here for more
information.
To access your Seurat object assay(s):
so@assaysTo access your Seurat object reductions:
so@reductionsTo access your Seurat object meta.data:
so@meta.data
# Or, to just display the first n rows of meta.data:
head(so@meta.data, n = 10)The column names in your meta.data will correspond to
the variables and latent variables you
see in the Pluto app. You will also see additional columns that were
added during the preprocess workflow, mostly related to QC. If you are
reading in a final Seurat object, you will
see columns that correspond to your cluster annotation sets.
To get a list of available meta.data columns in your
Seurat object:
colnames(so@meta.data)Visualization using the Seurat package
Once a Seurat object has been downloaded from Pluto, we
can use a variety of base plotting functions from the
Seurat package. We can further modify and customize these
plots using ggplot2.
Dimensional reduction plot
Using the Seurat DimPlot() function, make a
scatter plot in UMAP space grouping cells by custom annotation set
“Default cluster - res. 0.1” (here, we can use one of our custom color
palettes that we defined in the Pluto app):
DimPlot(so, # the Seurat object
reduction = "umap", # could also be "tsne" or "pca"
group.by = "default_clusters_res_0_1", # custom annotation set
cols = custom_palettes$default_clusters_res_0_1, # custom color palette
pt.size = 0.1
) + # point size for plotting
labs(
title = "Default clusters - res. 0.1", # update plot title
x = "UMAP 1", # update x-axis label
y = "UMAP 2"
) # update y-axis label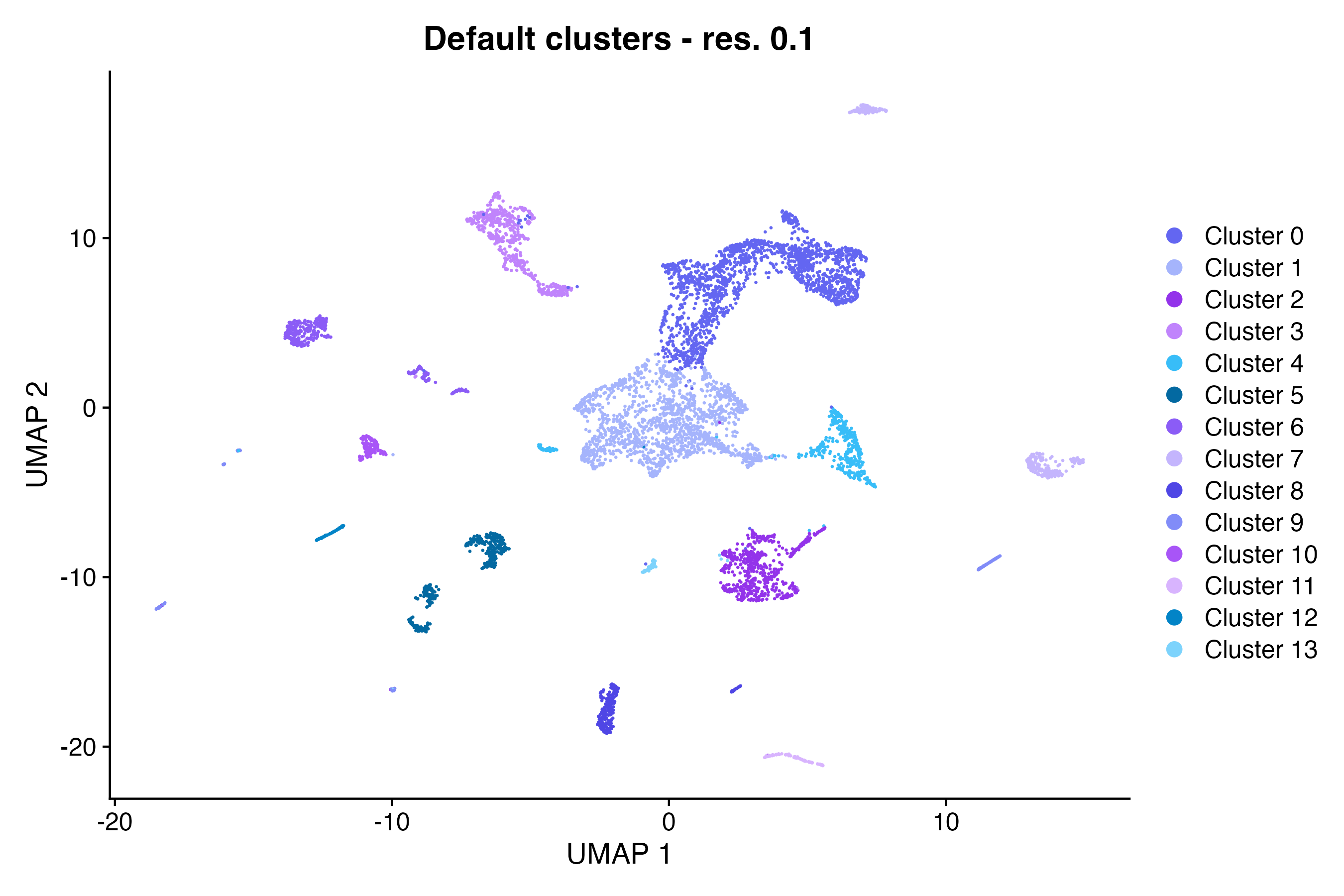
Using the Seurat DimPlot() function, make a
scatter plot in UMAP space grouping cells by timepoint:
# Create a custom color palette to group cells by timepoint
timepoint_cols <- c("#14b8a6", "#fcd34d", "#ef4444")
names(timepoint_cols) <- c("24hpf", "36hpf", "48hpf")
DimPlot(so, # the Seurat object
reduction = "umap", # could also be "tsne" or "pca"
group.by = "timepoint", # available column in our meta.data, from our Pluto sample data
cols = timepoint_cols,
pt.size = 0.1
) + # point size for plotting
labs(
title = "Timepoint", # update plot title
x = "UMAP 1", # update x-axis label
y = "UMAP 2"
) # update y-axis label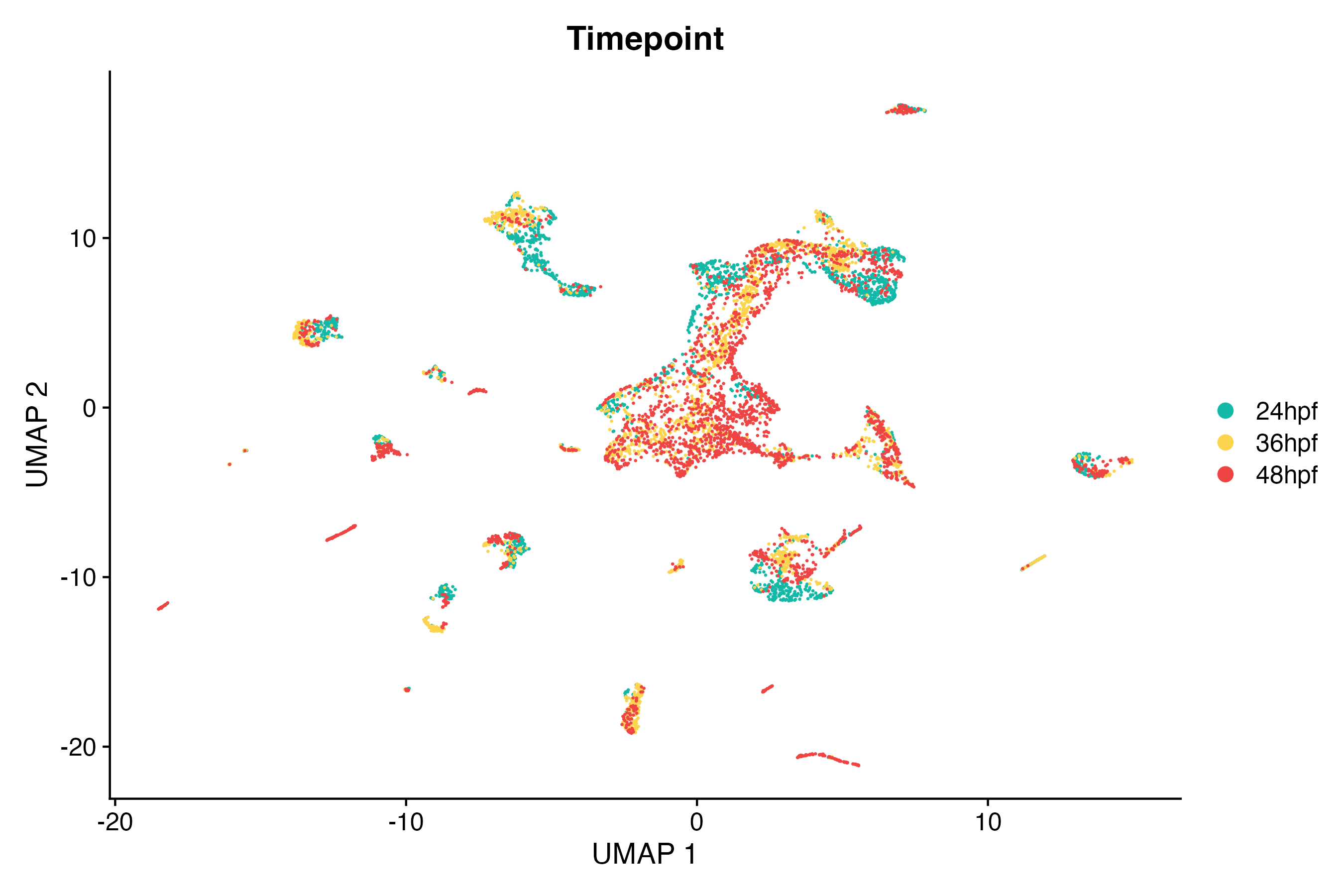
Using the Seurat DimPlot() function, make a
scatter plot in UMAP space grouping cells by custom annotation set
“Default cluster - res. 0.1” and splitting cells by timepoint:
DimPlot(so, # the Seurat object
reduction = "umap", # could also be "tsne" or "pca"
group.by = "default_clusters_res_0_1", # custom annotation set
cols = custom_palettes$default_clusters_res_0_1, # custom color palette
split.by = "timepoint", # custom annotation set
pt.size = 0.1
) + # point size for plotting
labs(
title = "Default clusters - res. 0.1", # update plot title
x = "UMAP 1", # update x-axis label
y = "UMAP 2"
) # update y-axis label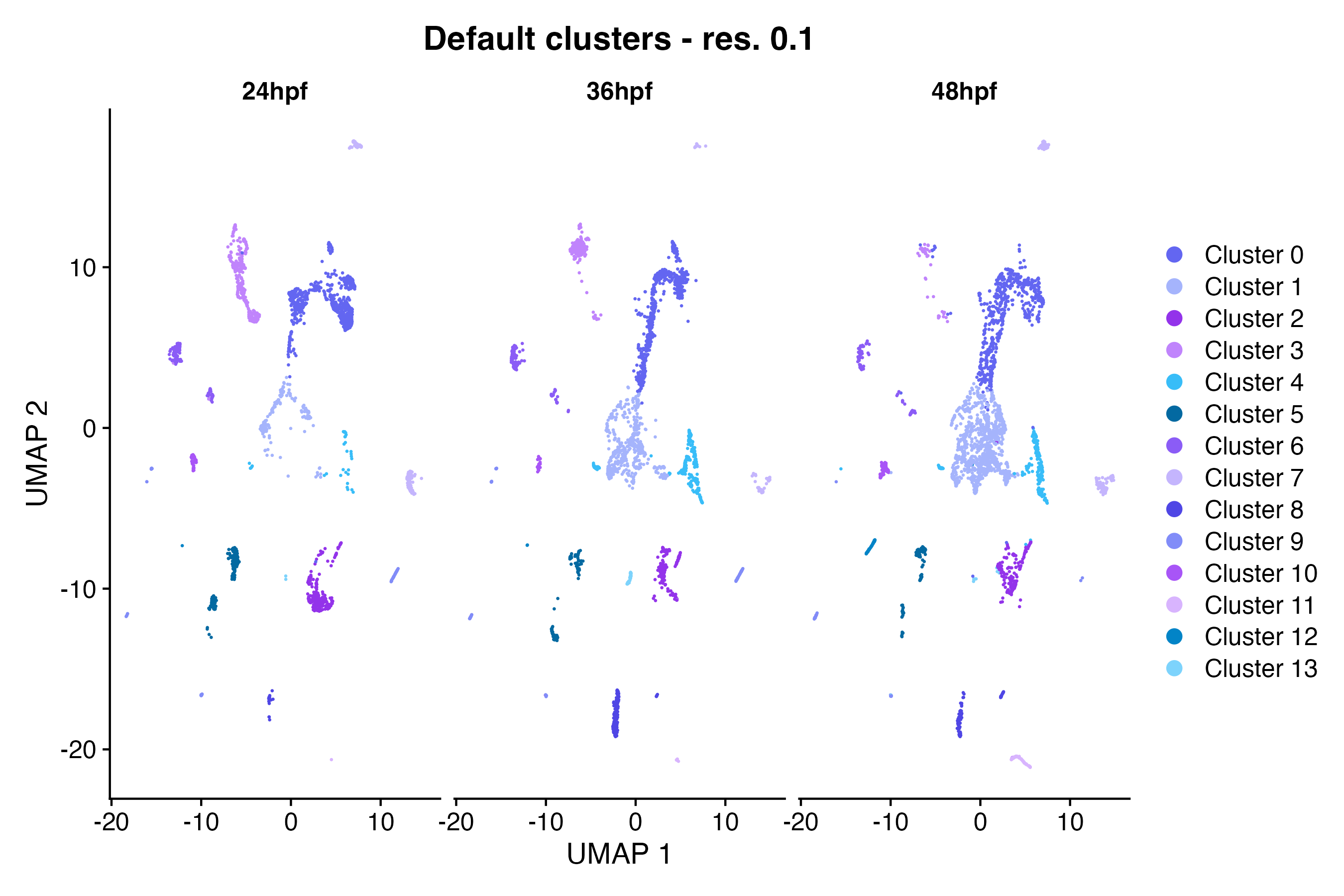
Marker feature expression
Violin plot
Using the Seurat VlnPlot() function, make a
violin plot grouping cells by custom annotation set “Default cluster -
res. 0.1”:
VlnPlot(so, # the Seurat object
features = c("olig2"), # feature(s) of interest
group.by = "default_clusters_res_0_1", # custom annotation set
cols = custom_palettes$default_clusters_res_0_1
) + # custom color palette
labs(
title = "Expression of olig2", # update plot title
x = "Default clusters - res. 0.1"
) # update x-axis label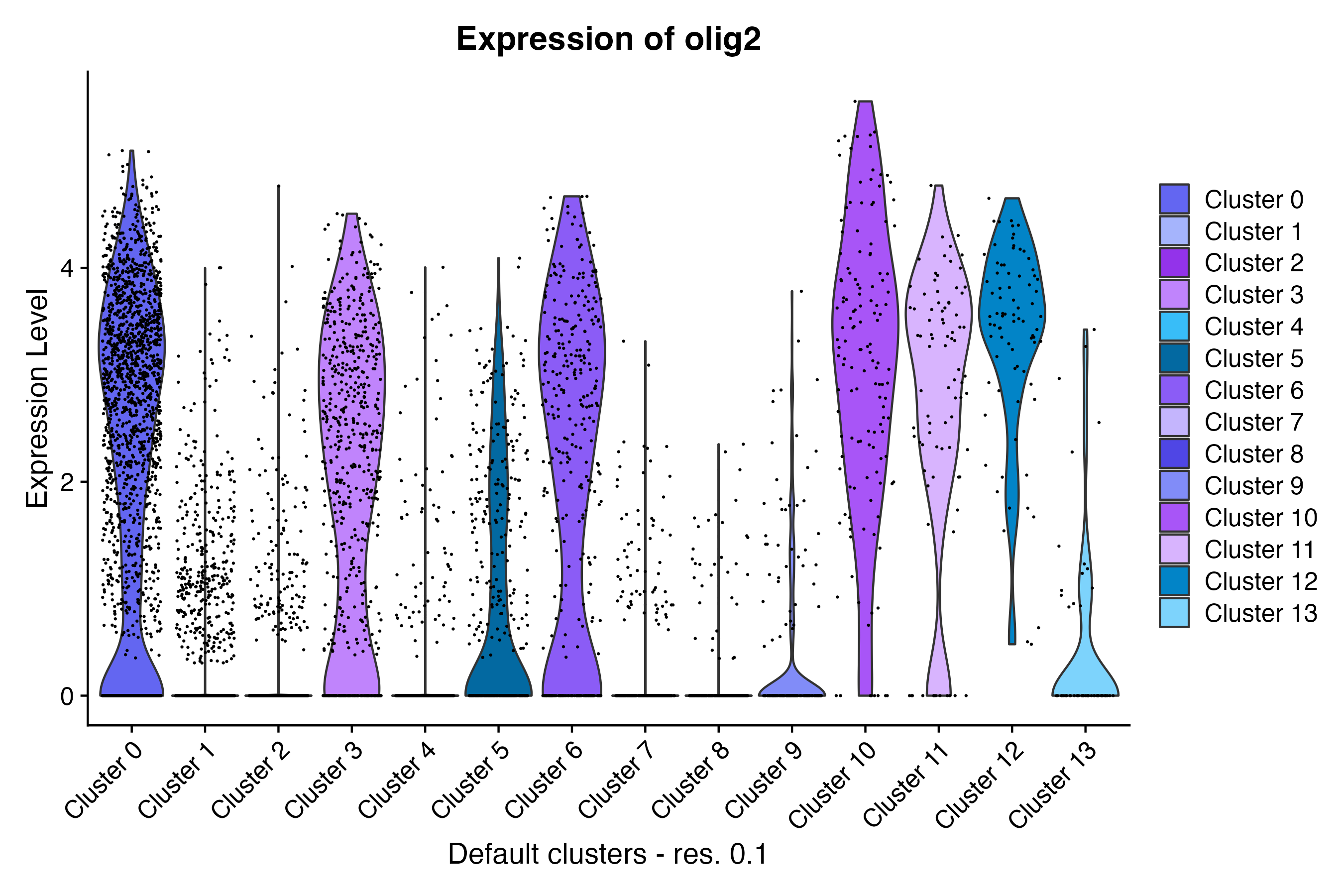
Ridge plot
Using the Seurat RidgePlot() function, make
a ridge plot grouping cells by custom annotation set “Default cluster -
res. 0.1”:
RidgePlot(so, # the Seurat object
features = c("olig2"), # feature(s) of interest
group.by = "default_clusters_res_0_1", # custom annotation set
cols = custom_palettes$default_clusters_res_0_1
) + # custom color palette
labs(
title = "Expression of olig2", # update plot title
y = "Default clusters - res. 0.1"
) + # update x-axis label
theme_ridges(center = TRUE) # center the x and y axis labels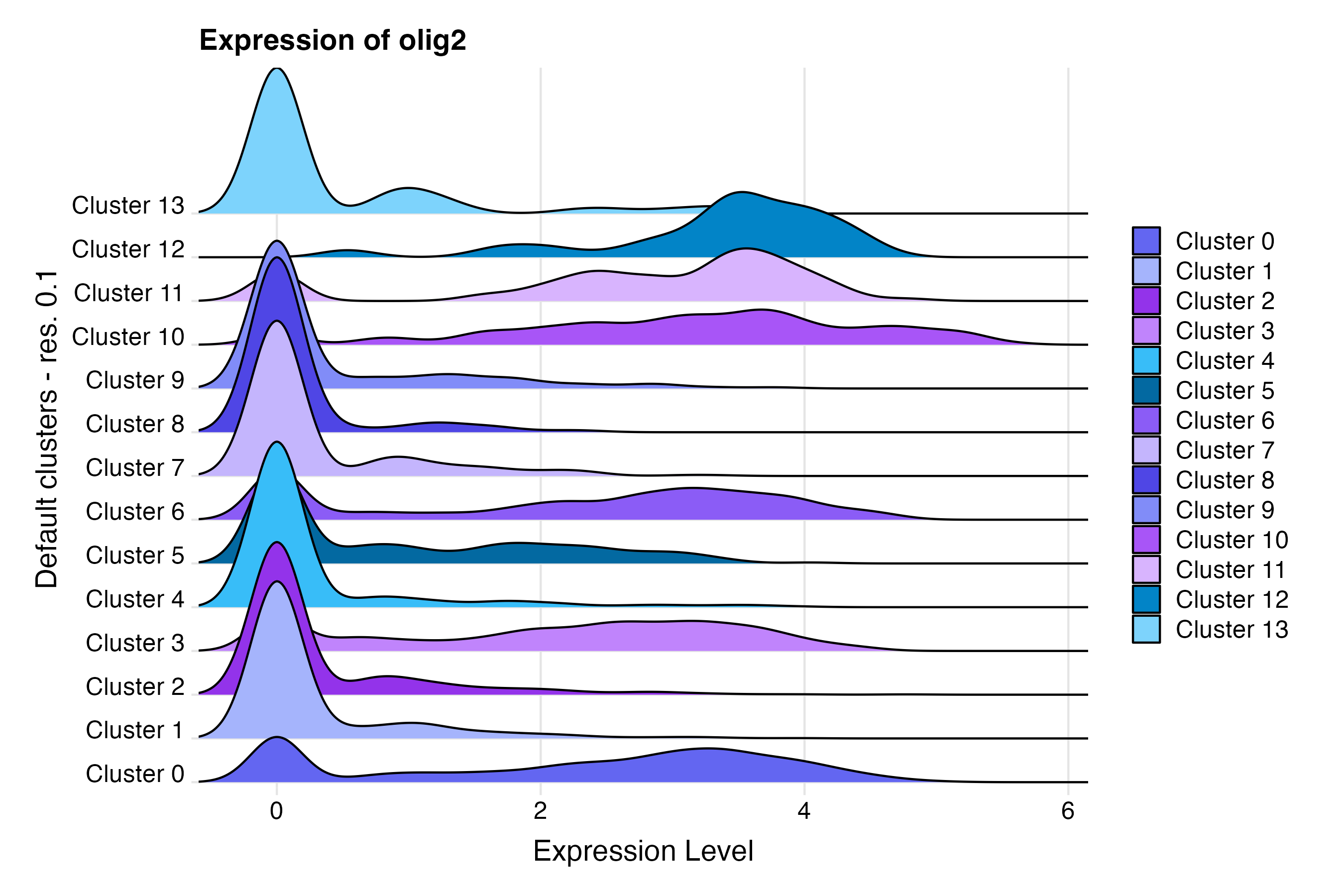
Feature plot
Using the Seurat FeaturePlot() function,
make a scatter plot in UMAP space colored by feature expression:
FeaturePlot(so, # the Seurat object
features = c("olig2"), # feature(s) of interest
reduction = "umap", # could also be "tsne" or "pca"
pt.size = 0.1
) + # point size for plotting
labs(
title = "Expression of olig2", # update plot title
x = "UMAP 1", # update x-axis label
y = "UMAP 2"
) # update y-axis label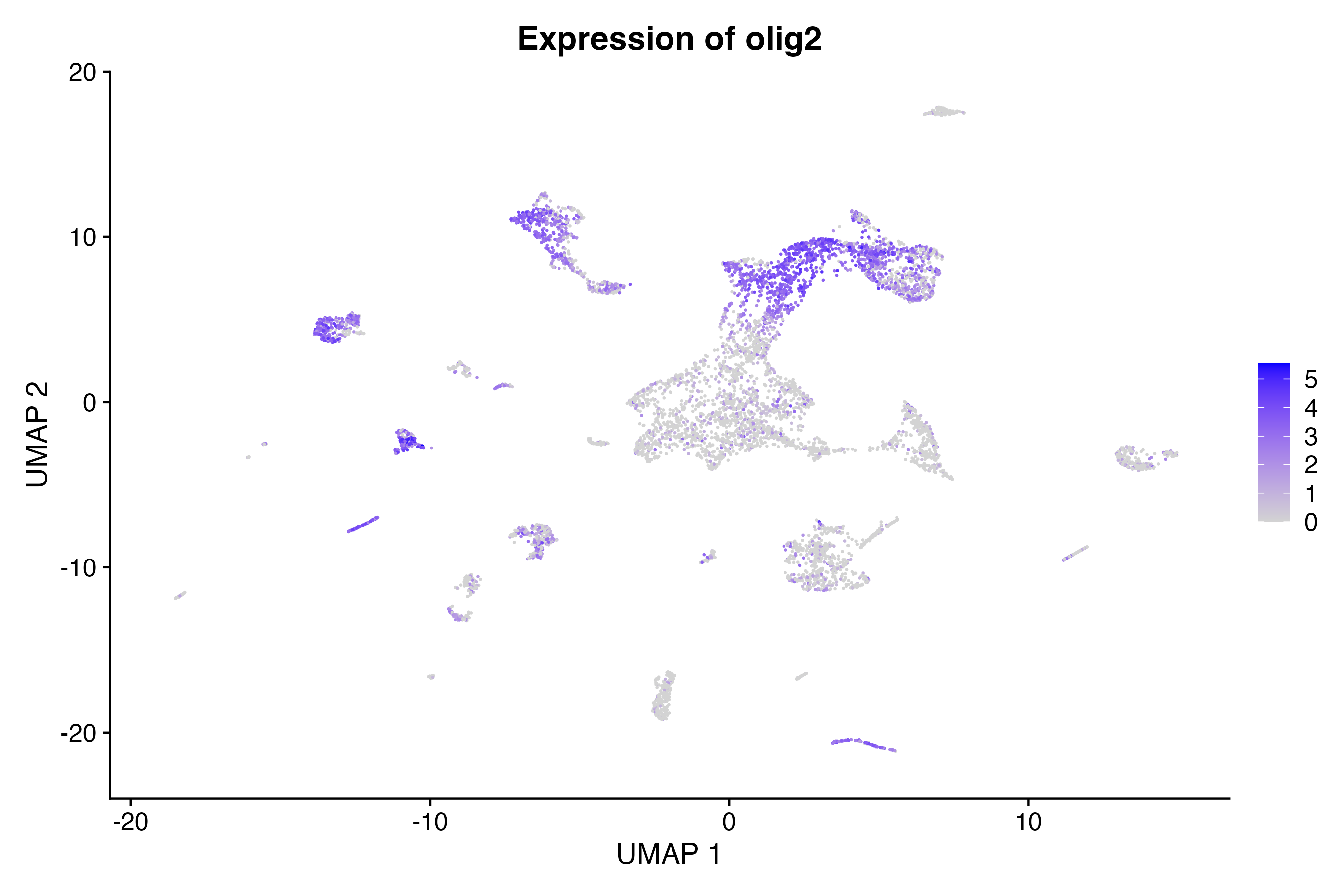
Dot plot
Using the Seurat DotPlot() function, make a
dot plot colored by feature expression:
DotPlot(so, # the Seurat object
features = c("olig2", "sox19a", "gfap", "dlb", "nkx2.2a"), # feature(s) of interest
group.by = "default_clusters_res_0_1"
) + # custom annotation set
labs(
title = "Key marker genes", # update plot title
x = "Genes", # update x-axis label
y = "Default clusters - res. 0.1"
) # update y-axis label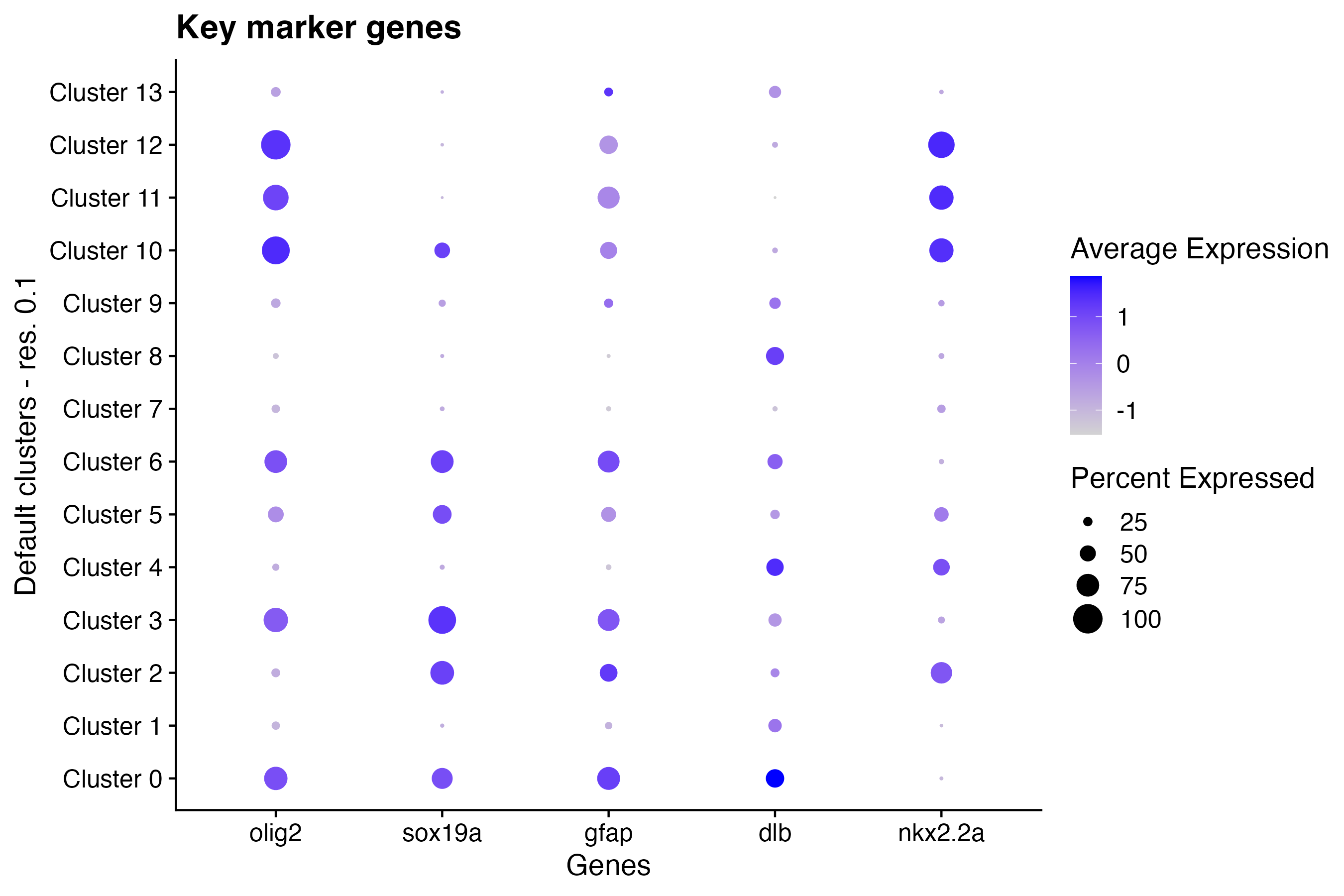
For more visualization tutorials using the Seurat
package, check out the Seurat visualization vignette here.